The Missing Kingdom: Why Fungi Must Be Central to Conservation Strategy
28 December 2025
Published online 15 September 2014
© Nature Biotechnology / Zhenqiang Su
Techniques such as high-throughput transcriptome and exome sequencing provide a wealth of data about the dynamics of gene expression, but the random nature of RNA sampling methods produce noisy measurements and recent analyses suggest that the data obtained so far are not entirely accurate.
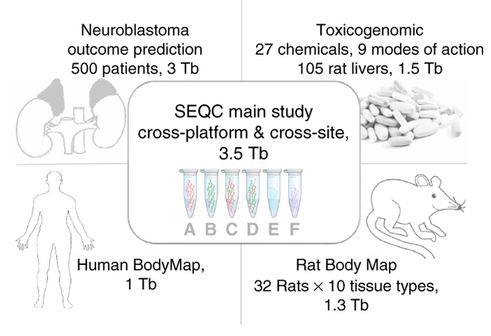
The Sequence Quality Control Consortium aims to assess the accuracy and reproducibility of RNA sequencing data, and the researchers involved have now published the primary results of the project.
They sequenced commercially available reference RNA samples at multiple laboratories using multiple sequencing platforms, to produce a data set comprising more than 100 billion nucleotides obtained in approximately 30 billion sequence reads, and compared the sequences with information from microarray and quantitative polymerase chain reaction (qPCR) analyses.
While measurements of relative gene expression are accurate and reproducible across multiple sequencing platforms, those of the absolute expression level of many genes differ according to the sequencing method.
The study also identified the vast majority of the approximately 55,000 known genes in the human genome, and around 2.6 million uncharacterised splice junctions, the sites where DNA is cut to remove non-coding regions and bring coding regions together, before it is transcribed into RNA.
Genes have multiple coding regions that can be spliced together in different ways, and most, if not all, have multiple ‘splice variants,’ but little is known about how they are produced. Better characterisation of these splice junctions may therefore provide insights into this process.
doi:10.1038/nmiddleeast.2014.223
28 December 2025
24 December 2025
24 December 2025
Sign-up to receive our e-alert update every two weeks to keep up with everything new on the portal.
Sign up for e-alerts
Stay connected: